| Version 7 (modified by , 14 years ago) (diff) |
|---|
MCF Data Model
The main goal of our model is to unambiguously specify a bioinformatics workflow execution in a distributed computational environment. The model includes the entire workflow specification and specifications for individual workflow tasks. Hence, we divide our model into two structural layers or interfaces. These are a job interface to specify the workflow and tool interface to specify an individual workflow task. These interfaces are used in two main framework use scenarios:
- specifying a workflow or job for execution, and
- adding a new tool to the framework tool repository. Hence, analyses provided by the tool can be included
into workflows.
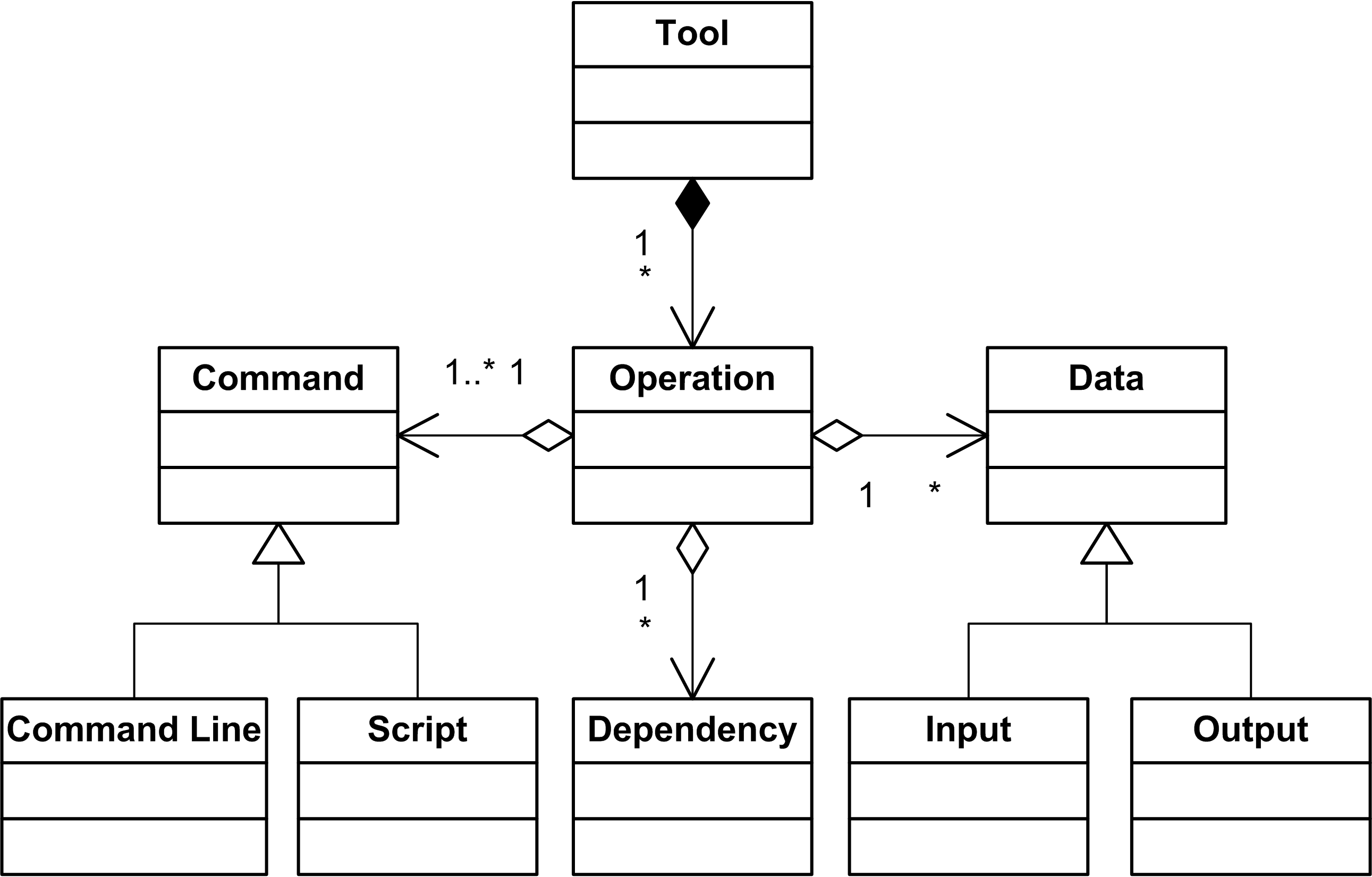
Let us examine these models in more detail. The tool model is similar to the Galaxy tool file, where a tool is described as a set of operations, which it can perform. A tool operation can participate in workflow execution. Our platform is aimed to support specifying any external analysis tool, which can be invoked from a command line or be run as an executable script (e.g. a shell or R-script). In contrast with the Galaxy description, we do not have complex operation parameters in the model. Instead, we treat an operation with differ- ent parameters as a set of separate operations. A list of tool operations grows depending on the complexity of the operations parameterisation. Still, the specification of an individual operation remains simple.

There are a number of reasons to keep the description of a single operation as simple as possible. Firstly, bioinformaticians typically use only a subset of available tool features in the majority of analysis workflows. Moving towards simpler operations importantly lowers the bar for bench biologists to be comfortable running operations without the need of massive bioinformatics background knowledge. Secondly, it is possible that a tool will be added to the framework by a person, who is not an expert in all operation parameters, but only interested in a particular execution scenario. In this case, it would be easy to add a short operation specification than an exhaustive list of all operation parameters. Furthermore, additional tool parameters can be added later as a new operation. All parameters defined in the tool can be reused by new operation definitions in good spirit of the Don’t Repeat Yourself (DRY) principle. Inputs, outputs and dependencies of a tool operation are specified explicitly to assure the correctness of workflow and provide means for dynamic error handling during workflow execution.
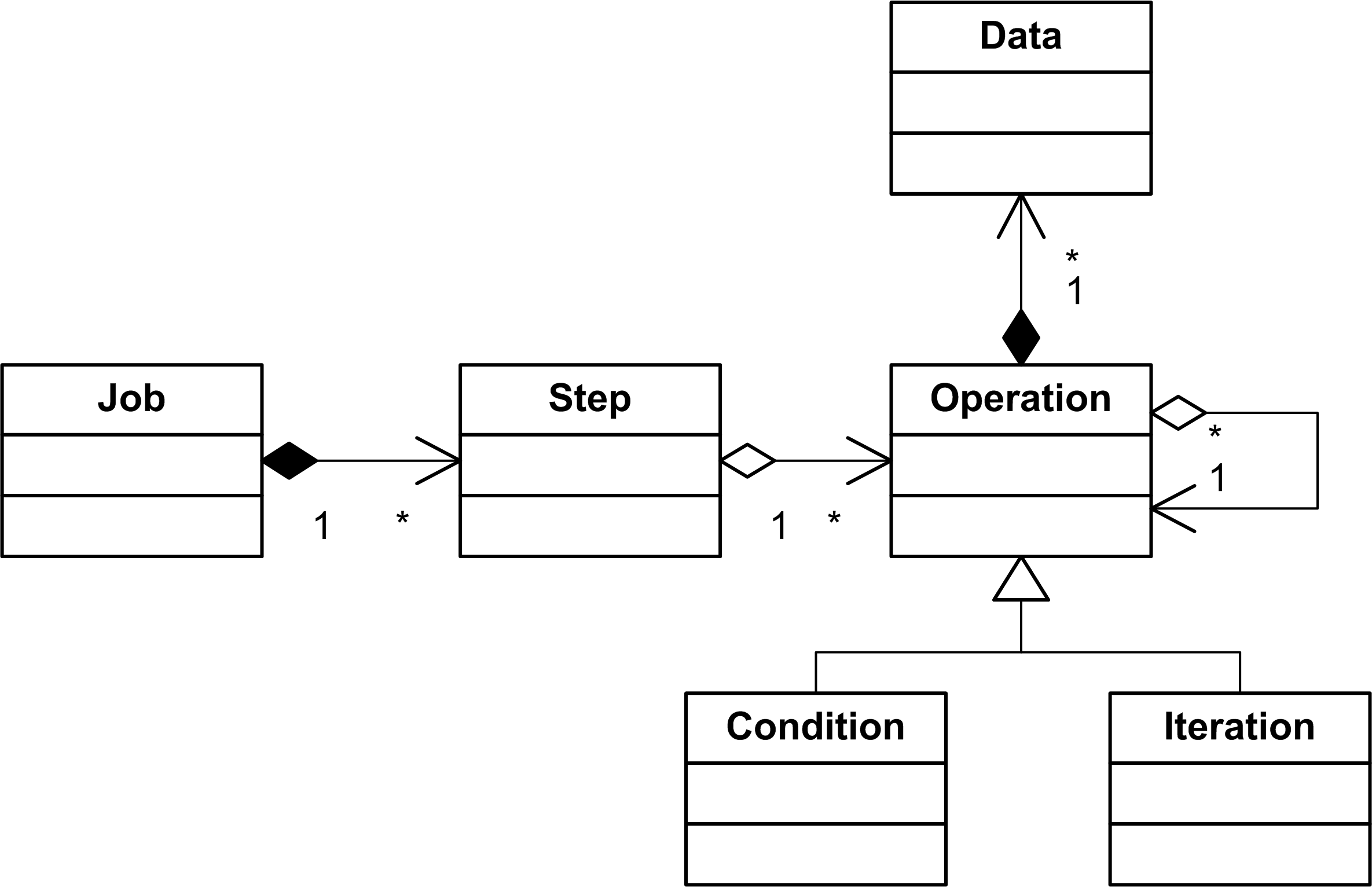
The second part of our model is the job model. It describes a workflow as a sequence of steps. A step consists of a set of operations. Operations of one step are executed in parallel and do not depend on each other. An operation can be a condition to specify that this operation should be executed under some conditions, which are results of the previous workflow step. The condition object class is present in the model, but it was not used, so far, in the designed workflows. Additionally, an operation can be an iteration to specify repeating of the same operation for different data (e.g. repeat the same operation for all chromosomes).

In the job model, the operation object is a concrete instance of a tool operation from the tool model. We would like to treat our computational environment as a cloud. Computational clouds are typically divided on segments. Transferring of large amounts of data between cloud segments can easily become a bottleneck of a workflow execution. Hence, computations should be performed close to the data storage. However, as practice shows, transferring of gigabytes of data from a single machine to a computational cluster is not a problem if one intends to run computationally intensive tasks for several days.
Attachments (2)
- tool.png (123.3 KB) - added by 14 years ago.
- job.png (97.2 KB) - added by 14 years ago.
{kind=link}
{kind=link}
Download all attachments as: .zip